
テュフズードインフォサービス
テュフズードインフォサービス

| 2022年12月の情報 |
欧州規制関連情報
EU 保健政策担当委員による MDR 移行期限延長の提言について
2022年 12月 9日に EU 保健政策担当委員より、EPSCO 評議会にて、MDR 移行期限延長の提言がなされましたのでご紹介します。
解説:
EU 政策担当委員(The Commissioners)とは 27名から成る欧州委員会の構成メンバーのことで、大統領によって特定の政策分野の責任を割り当てられています。その中で、保健及び食品安全政策を担う Commissioner である Stella Kyriakide 氏より、EU における雇用、社会政策、健康および消費者問題に関する評議会(EPSCO)の開会の辞の中で、高リスクの医療機器は 2027 年、中リスクおよび低リスクの医療機器は 2028 年という、新しい移行期限の提言がなされました。
そして、この提言の正当性について、MDD に基づいて発行された約 23,000 の EC 認証書の対象となる医療機器が未だに MDR への移行が完了しておらず、2024年 5月 26日に期限切れとなることが予想されるため、EU が医療機器不足に陥るリスクに直面しているためであると述べています。
本提言は、下記欧州委員会の Web ページにて閲覧可能な公式なものですが、その施行までには欧州議会での採決を始めとした数々の手続きが必要とされており、修正の可能性もあります。引き続き EU の動向を注視する必要がありますが、少なくとも、この提言が MDR への移行手続きをゆっくり進めることを許容する理由には決してなりえないと、欧州では理解されています。
参照 URL:
https://ec.europa.eu/commission/presscorner/detail/en/SPEECH_22_7627
関連セミナー:
欧州医療機器規則(MD REGULATION)改正対策勉強会
欧州 IVD 規則(IVD REGULATION)改正対策勉強会
特別講演 欧州医療機器規則 MDR 最新情報と医療機器の臨床評価
スイス規制関連情報
米国 FDA が承認・認可した医療機器を受け入れることを可能にするためにスイス国内法の修正指示に対するスイス連邦議会の可決について
2022年 11月 28日にスイス連邦議会は、米国 FDA が承認・認可する医療機器を受け入れることを可能にする国内法の適応指示案を可決しましたのでご紹介します。
解説:
2001年以来、スイス国内において医療提供者と患者は、CE マーキングを貼付した医療機器製品しか利用することができませんでした。しかし、スイス連邦議会の下院は、スイス連邦評議会に対し、従来の CE マーキング付きの機器と並行して、新たに、米国 FDA が承認・認可した医療機器をスイス市場へ上市することを許容するために、関連するスイス国内法を修正するよう指示する決定を下しました。
これは、ダミアン ミュラー国家評議員の動議「スイス国民に供給するための医療機器の調達における選択肢の余地を増やす」(20.3211)に基づいたもので、上院では、今年 5月 30日に承認されていました。
EU の MDR が医療機器メーカーが克服しなければならない規制上のハードルを高めたため、スイスでは、多くのスタートアップ企業や中小企業が医療機器の初期承認を、米国 FDA に求めるようになったと言われています。スイスの医療機器業界団体である Swiss Medtech は、この可決を支持する声明を発表しました。
なお、この動機が施行されるまで踏むべき 16 の手続きのうち、現在は、まだ最初の手続きの段階にあることを鑑みると、米国 FDA が承認・認可した医療機器をスイス市場へ上市できるまで、しばらく時間を要するとスイスでは理解されています。
参照 URL:
https://www.swiss-medtech.ch/en/news/politicians-decide-favour-patient-care
関連セミナー:
スイス医療機器関連規制セミナー
欧州規制関連情報
Team-NB Position Papers の発行について
2022年 10月に医療機器 ノーティファイドボディ協会(Team-NB)より、幾つかのポジションペーパーが発行されましたのでご紹介します。
解説:
Team-NB Position Papers の使用は MDR/IVDR 適合性を示す上で強制ではありませんが、異なるノーティファイドボディ(NB)間の適合性評価の平準化を促し、NBによる適合性評価に臨む製造業者が一読するに値するガイダンス文書です。発行された主なポジションペーパーは、次の通りです。
Best Practice Guidance for the Submission of Technical Documentation under Annex II and III of Medical Device Regulation (EU) 2017/745
NB へ提出する技術文書(Technical Documentation) の重要事項に関する一般的な指針を示しており、NB が期待している最低限の一般的レベルを規定しています。
Transfer Agreement
製造業者がある NB から別の NB へ自発的に変更する場合の MDR/IVDR に基づく変更条件が整理して記載されています。NB を変更する場合のテンプレートとして利用することも可能です。
Class D measures in the absence of EU Reference Laboratories
EU 標準試験所が存在しない場合の、クラスD IVD機器の検証のための措置が明記されています。一部のクラス D IVD機器では、PSUR(定期的安全更新報告)の作成頻度が 6 か月に一回へと高くなる可能性があると記されています。
Cybersecurity
製造業者がサイバーセキュリティの脅威に対応するための推奨事項として、IEC 81001-5-1 などの国際規格への適合、STRIDE などの体系的な脅威モデリング手法の採用、ISO 17025 認定試験所による検証とバリデーションのためのペネトレーションテストの実施、サイバーセキュリティ PMS(市販後監視)が挙げられています。
'Off-Label’ Use of a device under the EU Medical Device Regulation 2017/745
「Systematic off-label use」(認証された医療機器の意図した目的および適応から外れたところで、繰り返しまたは継続的に使用されることを意味する)、および「Unmet medical need」(機器が非体系的に無作為に使用されたことを意味する)の言葉の定義が記載されています。製造業者は機器の誤使用を減らすための措置を講じる必要がありますが、この新たに特定された使用をカバーする必要があるかどうかを評価することも期待されると述べられています。
参照 URL:
https://www.team-nb.org/team-nb-position-paper-on-bpg-technical-documentation/
https://www.team-nb.org/team-nb-position-paper-on-transfer-agreement/
https://www.team-nb.org/class-d-measures-in-the-absence-of-eu-reference-laboratories/
https://www.team-nb.org/cyber-security-position-paper/
https://www.team-nb.org/data-generated-from-off-label-use-of-a-device-under-the-eu-medical-device-regulation-2017-745/
関連セミナー:
欧州 MD 規則 (MD Regulation) 改正対策勉強会
欧州 IVD 規則 (IVD Regulation) 改正対策勉強会
欧州規制関連情報
欧州議会での Corporate Sustainability Reporting Directive (CSRD) の採択について
2022年 11月 10日に欧州議会において、新たな欧州指令 Corporate Sustainability Reporting Directive (CSRD) が採択されましたのでご紹介します。
解説:
CSRD は、企業の社会的および環境的影響に関する持続可能性に関する情報を定期的に開示することを義務付けた欧州指令です。欧州委員会は、2023 年 6 月までに最初の一連の基準を採用する予定です。適用される欧州企業は、2024 年から 2028 年の間に 49,000 社にものぼると推測されています。また、欧州域外の企業であっても、欧州域内で 1.5億ユーロ以上の売上があり、欧州域内に少なくとも一つの子会社、あるいは支店を持つ全ての企業にも適用されます。適用される企業は、環境、社会、ガバナンス(ESG)への影響に関する報告書を提供する公的な説明責任を負う必要があるとされています。
参照 URL:
https://www.europarl.europa.eu/news/en/press-room/20221107IPR49611/sustainable-economy-parliament-adopts-new-reporting-rules-for-multinationals
関連セミナー:
医療機器関連規制視点のサプライチェーンマネジメント(SCM)基礎力養成セミナー
| 2022年 11月の情報 |
行政当局のリストの更新について
2022年 10月に欧州委員会より、更新版行政当局リストが発行されましたのでご紹介します。
解説:
欧州加盟国の行政当局について、以下それぞれの業務の連絡窓口の最新版一覧表が欧州委員会より発行されました。
欧州医療機器規則(MDR)および欧州体外診断用医療機器規則(IVDR)は、製造業者に対して、例えば、公衆の健康への脅威がある場合という情報を得た場合は、2日以内に行政当局へ報告することを確実とするための手順を文書化することを要求しています。欧州加盟国それぞれのビジランス連絡先は、この最新版一覧表より確認することが可能です。
参照 URL:
https://health.ec.europa.eu/medical-devices-sector/new-regulations/contacts_en
関連セミナー:
欧州医療機器規則(MD REGULATION)改正対策勉強会
欧州 IVD 規則(IVD REGULATION)改正対策勉強会
※ 次回、2023年 3月の開催を予定しています。
欧州 IVD 規則(IVD REGULATION)アドバンスド・セミナー
※ 次回、2023年 7月の開催を予定しています。
英国医療機器規則 の経過措置期間延長について
2022年 10月 25日に、英国医薬品庁(Medicines and Healthcare products Regulatory Agency: MHRA)より、医療機器規則 Medical Device Regulations 2002 (UK MDR 2002) の実施猶予期間を 12か月延長する旨の決定がなされましたのでご紹介します。
解説:
UK MDR 2002は、2023年 6月 30日までの間、CEマーキングを貼付した医療機器製品を英国市場において受け入れることが決定しています。この経過措置について、MHRAから英国政府の諮問に対し、英国内におけるシステムの準備態勢と医療機器供給途絶のリスクを最小化するためのアプローチを確保する必要があるとの回答が、2022年 6月 26日に政府から出されました。
今回の決定は、当初の経過措置期間を12か月延長し、2024年7月1日からUK MDR 2002の適用開始を目指すこととしたものです。この決定により、CEマークを貼付した医療機器製品は2024年 6月 30日までの間、引き続き英国市場に流通することができます(2024年 7月からは、英国の UKCAマークの表示を義務化)。
また、2023年春までに法案を提出し、今回の経過措置と新たな市販後調査の要件について発効させる予定であることを表明しています。
参照 URL:
https://www.gov.uk/government/publications/implementation-of-the-future-regulation-of-medical-devices-and-extension-of-standstill-period
| 2022年 10月の情報 |
医療機器 510(k) 用の電子提出テンプレート、及び食品医薬品化粧品法の第522項に基づく市販後監視に関するガイダンス文書の発行について
米国 FDA(食品医薬品局)より、2022年 9月 22日に最終版医療機器 510(k) 用の電子提出テンプレートが、同年 10月 7日に食品医薬品化粧品法 522項に基づく市販後監視に関する最終版ガイダンス文書が発行されましたのでご紹介します。
解説:
医療機器 510(k) 用の電子提出テンプレートは、電子形式による市販前通知(510(k))提出の追加基準、これらの基準を確立するためのタイムテーブル、法定要件からの免除基準などを規定してします。FDA は、2023 年 10 月 1 日を、510(k) の電子提出の施行期日しています。郵送で提出された 510(k) 申請が2023 年 10 月 1 日より前にFDA によって受領された場合は、その申請をFDAは受け入れる意向のようです。
今回公示された市販後監視に関するガイダンス文書は、2016年 5月に発行された旧ガイダンス文書を置き換える位置づけにあります。食品医薬品化粧品法の第522項は、FDAに対して、Class III の機器への承認、或いは Class II の機器へのクリアランス、またはその後の任意の時点で、製造業者に市販後調査を実施することを要求する権限を与えています。このガイダンス文書は、製造業者が、市販後監視命令(いわゆる522 orders)で規定された義務を履行するために、どのように、市販された機器に関するデータまたはその他の情報の積極的、体系的、科学的に有効な収集、分析、および解釈するべきかの方針を規定しています。
参考:文書のダウンロード
Electronic Submission Template for Medical Device 510(k) Submissions [PDF]
Postmarket Surveillance Under Section 522 of the Federal Food, Drug, and Cosmetic Act [PDF]
関連セミナー:
FDA 規制 510(k) 申請方法解説セミナー
※ 次回、2023年9月の開催を予定しています。
欧州規制関連情報(スイス)
スイスの医療機器データベース(Swissdamed)について
2022年 9月 13日に、スイス医薬品局(Swissmedic)より、医療機器データベース(Swissdamed)に関する公示がありましたのでご紹介します。
解説:
スイス連邦と EU の間で医療機器適合性評価の相互承認に関する合意に至っていないため、エコノミックオペレータと医療機器固有識別(UDI)の登録は Swissmedic によって管理されます。そして、これらを登録するための新しいデータベース(Swissdamed)の概要が公示されました。
Swissdamedは、欧州のデータベース(EUDAMED)に類似した設計であるとも言われていますが、次のような違いが明示されています。
元の登録情報は Swissdamed にロールバックされるため、既に Swissmedic に登録済みの事業者は、再度登録する必要がないと記載されています。さらに、この登録手続きは、スイス医療機器政令(MedDO)及び体外診断用医療機器政令(IvDO)における関連改正があった後にのみ、義務化されると明記されています。
参照 URL:
https://www.swissmedic.ch/swissmedic/en/home/medical-devices/medizinprodukte-datenbank.html
関連セミナー:
スイス医療機器関連規制セミナー
スイス体外診断用医療機器関連規制セミナー
厚生労働省関連情報:
令和 4年 9月 30日に、医療機器のユーザビリティエンジニアリングに関わる通知が出されましたのでご紹介します。
解説:
本通知の発出により、IEC 62366-1 に相当する JIS規格である JIS T 62366-1 の日本の規制上の取り扱いが変わり、経過措置期間終了日(令和 6年 3月 31日)の後、基本要件基準 第9条、第16条への適合を示す上で、JIS T 62366-1:2022(IEC 62366-1:2015 + AMD1:2020)を適用規格とすることが事実上必須とされたものと考えられます。以下は、本通知の内容の抜粋となります。
参考:文書のダウンロード
薬生機審発 0930 第1号
薬生監麻発 0930 第1号
令和 4年 9月 30日
医療機器のユーザビリティエンジニアリングに係る要求事項に関する日本産業規格の改正の取扱いについて [PDF]
令和4年 9月 13日に発出された、医療機器の包装へのバーコード表示(UDI及び注意事項等情報)に関わる通知、事務連絡をご紹介します。
解説:
改正医薬品医療機器法において、医薬品、医療機器の包装にバーコードを表示し、アプリをインストールしたスマートフォン等で添付文書・関連文書に記載された注意事項等情報に電子的にアクセスできるようにすることが義務付けられています。本件に関連する通知と事務連絡が、令和4年 9月 13日に出されています。これまでに出された通知、事務連絡を含めて、以下に整理します。
期日(2023年 8月)に間に合うように、表示すべきバーコードの手配、製品表示の切り替え等を、計画的に準備していただく必要がありますのでご注意ください。
通知:
令和3年 2月 19日
薬生安発0219第1号
医薬品等の注意事項等情報の提供について
※令和3年に上記の通知が出されていたが、一部改正版が令和4年 9月 13日に出された。
参考:文書のダウンロード
令和4年9月13日
薬生安発0913第5号
「医薬品等の注意事項等情報の提供について」の一部改正について [PDF]
※改正前の内容もこのファイルで確認できます。
事務連絡:
令和3年2月19日
事務連絡
「医薬品等の注意事項情報提供について」に関する質疑応答集 (Q&A)について
※上記の一部改正版が令和3年6月11日に出された。
令和3年6月11日
事務連絡
「医薬品等の注意事項等情報の提供について」に関する質疑応答集(Q&A)の一部改正について
※上記の一部改正版が令和3年7月14日に出された。
令和3年7月14日
事務連絡
「医薬品等の注意事項等情報の提供について」に関する質疑応答集(Q&A)の一部改正について
※上記の一部改正版が令和4年9月13日に出された。
参考:文書のダウンロード
令和4年9月13日
事務連絡
「医薬品等の注意事項等情報の提供について」に関する質疑応答集(Q&A)の一部改正について [PDF]
あわせて「医療機器、体外診断用医薬品等 を特定 するための符号の容器への表示の方法(UDI)」についても通知と事務連絡が令和4年 9月 13日に出されていますのでご紹介します。
通知:
参考:文書のダウンロード
医政産情企発 0913 第1号
薬生安発 0913 第1号
令和4年 9月 13日
医療用医薬品を特定するための符号の容器への表示等について [PDF]
事務連絡:
参考:文書のダウンロード
令和4年 9月 13日
医療機器、体外診断用医薬品等を特定するための符号の容器への表示等に関する質疑応答集(Q&A)について [PDF]
関連セミナー:
医療機器製品へのバーコード表示の義務化に関する勉強会
※ 2022年12月22日の開催を予定しています。
| 2022年 9月の情報 |
米国規制関連情報
医療機器の EMCガイダンス文書改訂版の発行について
2022年 6月に米国 FDA より、医療機器の EMC ガイダンス文書改訂版が発行されましたのでご紹介します。
解説:
近年、医療機器において、RFID や NFC システムなどを利用した製品が増えつつあります。RFID や NFC システムなどは、無線通信でのデータ転送といった製品に新たな付加価値を与える一方で、妨害電磁エネルギー(ノイズ)放出という形で製品に対する新たなリスク源にもなりえることが知られています。
このガイダンス文書の改訂は、この新たなリスクに対して、これまでの FDA 認定合意規格(recognized consensus standards)では適切に対処できない可能性があることを FDA が認めたことがその背景にあります。米国の 510 (k) 申請において FDA 合意規格(recognized consensus standards)への適合を適合宣言によって示す場合、IEC 60601-1-2:2014(第4.0版)の使用は、2023年12月17日までしか認められなくなりました。その後は、IEC 60601-1-2:2020(第4.1版)のみを受理するとしています。
このガイダンス文書改訂版は、2020 年 11 月 17 日に発行された旧版ガイダンスに代わり、AIM 7351731、あるいは IEC 60601-1-2:2020 の箇条 8.11 を含むようにテストすることを推奨しています。医療機器メーカーは、少なくとも近接磁界イミュニティ試験に慣れ始める必要があると記載されています。
参照 URL:
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/electromagnetic-compatibility-emc-medical-devices
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/detail.cfm?standard__identification_no=41539
関連セミナー:
医療機器の EMCノイズ対策手法と IEC 60601-1-2 第4.1版に有効なメソッド
※ 次回、2023年 7月の開催を予定しています。
欧州規格関連情報
MDR 及び IVDR の下でのボーダーライン及びクラス分類マニュアルの初版発行について
2022年 9月に欧州委員会より、欧州医療機器規則(MDR)に基づく医療機器および欧州体外診断用医療機器規則(IVDR)に基づく体外診断用医療機器に関するボーダーライン及びクラス分類マニュアルの初版が発行されましたのでご紹介します。
解説:
MDR および IVDR の下で利用できる待望の新バージョンが発行されました。このマニュアルでいう「ボーダーライン」とは、ある製品が欧州規則上、医療機器(medical device)か、あるいは体外診断用医療機器(in vitro diagnostic medical devices)と見なされるか境界という意味です。
法的拘束力はありませんが、ある製品が医療機器、あるいは体外診断用医療機器とみなされるかどうかについて、事例を挙げて検討しています。該当する製品がある場合は参考になると思われます。旧マニュアルからの主な更新点、すなわち MDD 及び IVDD の下でのマニュアルと本マニュアルとの大きな変更点は、次の通りです。
※ 次回、2023年 3月の開催を予定しています。
欧州 IVD 規則(IVD REGULATION)アドバンスド・セミナー
※ 次回、2023年 7月の開催を予定しています。
| 2022年 8月の情報 |
欧州規格関連情報
MDR / IVDR 整合規格提案リスト更新版の公示
2022年 6月 1日に、欧州委員会のWEBページにおいて、 MDR / IVDR 整合規格提案リストの更新版が公示されましたのでご紹介します。
解説:
従来より MDR / IVDR 整合規格提案が公表されていましたが、2022年 6月 1日に、MDR / IVDR 整合規格提案リストの更新版が公示されました。この文書は提案書であり、記載された規格の整合規格化が公式に決定されたわけではありませんが、どのような規格が欧州整合規格になるか(あるいは、ならないか)を推測する上では有効な情報源となります。
例として、サイバーセキュリティ関連の情報を挙げてみます。
MDR 附属書 I(安全性と性能の要求事項)の 17.2項には、情報セキュリティを含めたリスクマネジメントという記載がありますが、それに対する欧州整合化規格はまだありません。そのような際に、今後の欧州整合規格の動きを推し量るために参考になるのが本提案書です。これまで公表されていた更新版提案リストでは、IEC TR 60601-4-5 が整合規格化を提案するリストに掲載されていましたが、2022年 6月 1日に公示された更新版提案リストでは、提案から除外する文書と位置付けられています。
これにより、IEC TR 60601-4-5が欧州整合化規格として認知される可能性は下がったと考えられています。
参照 URL:
https://ec.europa.eu/docsroom/documents/50274
関連セミナー:
医療機器のサイバーセキュリティ関連規制、規格動向
欧州規格関連情報
欧州医療機器規則(MDR)の附属書 XVI に規定された非医療機器の分類に関する実施細則提言案のパブコメ募集について
2022年 8月 11日に、欧州委員会より、MDR の附属書 XVI に規定された非医療機器分類に関する実施細則に関する提言案が発行されましたのでご紹介します。
解説:
MDR の定義上は医療機器ではないが、MDR の規制対象となる非医療機器が附属書 XVI のリストに掲載されています。今回、欧州委員会より新たに発表された機器分類に関する実施細則提言案には、例えば、次のような機器が明記されました。
この提案がこのまま通ると、上記に該当する非医療機器は、リスクに応じて分類され、それと同等の医療機器と同じ市販前及び市販後の要件が課せられることを意味しています。該当する非医療機器メーカーは、この提言案を読み、欧州委員会へ意見することが可能です。パブコメ(意見公募)の締め切りは、2022年 9月 8日とされています。
参照 URL:
https://ec.europa.eu/info/law/better-regulation/have-your-say/initiatives/12972-Medical-devices-reclassification-of-products-without-an-intended-medical-purpose_en
関連セミナー:
欧州医療機器規則(MD REGULATION)改正対策勉強会
医療機器への GS1 コードを記載義務について
医薬品医療機器総合機構(PMDA)のホームページに掲載された添付文書への、医療機器(容器・被包)上への GS1 コードの表示による紐づけが義務化されます。まだ猶予期間中ですが、期限に間に合うように GS1コードの用意、表示場所の決定、表示切替えの手配等の準備をしていただく必要がありますので、あらためてご紹介します。
解説:
2021年の本情報ページで掲載した通り、医療機器(※)の添付文書を、医薬品医療機器総合機構(PMDA)のホームページに掲載することが義務化されました。
※対象:全ての医療機器(クラス I - IV)。ただし、主として一般消費者の生活の用に供されることが目的とされる医療機器を除く(薬機法施行規則別表第4の2 及び 令和 3年厚生労働省告示第44号参照)
それに続いて、2年間の猶予期間を経て 2023年 8月 1日からは、医療機器(クラス I - IV)の容器又は被包への GS1 コードを表示することが義務化されます。それにより、使用者が、スマートフォン又はタブレット等を用いて医薬品医療機器総合機構(PMDA)のホームページにある最新版の添付文書へアクセスできるようになります。
なお、この GS1 コードは UDI 表示ではありません。上記の通り、最新の添付文書へのアクセスが目的となります。参考となる情報は、以下のリンクからご覧いただけます。
参照URL:
https://www.pmda.go.jp/safety/info-services/0003.html
参考:文書のダウンロード
薬生安発0219 第1号
令和3年 2月 19日
医薬品等の注意事項等情報の提供について [PDF]
| 2022年 7月の情報 |
欧州規制関連情報
欧州体外診断用医療機器規則(IVDR)Class D 機器における Common Specifications(CS)について
2022年 7月 4日に、欧州委員会より、欧州体外診断用医療機器規則(IVDR)における幾つかの Class D 機器に関する Common Specifications(CS)が記載された欧州官報が発行されましたのでご紹介します。
解説:
欧州委員会は、HIV 検査や SARS-CoV-2(新型コロナウィルス)検査など、いくつかのタイプの高リスクな体外診断用医療機器に関する CS を採用しました。さらに、欧州委員会は、これらの概要を示す新規 WEB ページを立ち上げました。この施行規則は、発行後から 20日目にあたる 2022年 7月 25日に発効されています。
ここでリスト化されている機器は、IVDR の附属書 VIII、Rule 1 に該当する機器、あるいは Rule 2 で規定されている血液型マーカーに該当します。製造業者が CS に準拠するための移行期間は、2024年 7月 25日までの 2年間とされています。
参照 URL:
https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32022R1107&qid=1657009507445
https://health.ec.europa.eu/vitro-diagnostics/common-specifications_en
関連セミナー:
欧州 IVD 規則(IVD REGULATION)改正対策勉強会
欧州 IVD 規則(IVD REGULATION)アドバンスド・セミナー
JIS 規格関連情報(パブリックコメント)
医療機器関連 JIS 規格のパブリックコメント募集について
医療機器関連 JIS 規格の制定案 2件、改正案 5件(下記)についてのパブリックコメント募集が 2022年 7月 15日に公示されました。医療機器メーカーにとって非常に影響の大きい規格も含まれていますのでご紹介します。
解説:
医療機器のソフトウェアを含むヘルスソフトウェアのサイバーセキュリティについては、 2021年 12月に IEC 81001-5-1:2021 という国際規格が発行されており、この IEC 81001-5-1 が今後、世界各国で医療機器法規制に取り入れられて用いられるものと予測されています。今回、IEC 81001-5-1 に基づいた JIS 規格案のパブリックコメントが公示されましたが、本 JIS 化により今後、日本でも JIS T81001-5-1(IEC 81001-5-1)が規制に取り込まれていく流れになっていく可能性があると思われます
JIS T0601-1 の改正案も注目されます。現在の JIS T0601-1 が 「IEC 60601-1:2005 + AMD1:2012」 を基にして制定されているところ 「IEC 60601-1:2005 + AMD1:2012 + AMD2:2020」を基にした規格にすることが、今回の改正案作成の目的となります。
新規制定案が提示された JIS T60601-1-6 は、国際規格 IEC 60601-1-6 に相当する能動医療機器を対象としたユーザビリティの JIS 規格となります。
現在、JIS T0601-1:2017 はユーザビリティに関して国際規格 IEC 60601-1-6 を引用規格おり(IEC 60601-1-6 に相当する JIS 規格が存在しないため)、更に IEC 60601-1-6 が IEC 62366-1:2015 + AMD1:2020 を引用規格とする形になっています。今回、JIS T0601-1 改正案とともに JIS T60601-1-6 制定案も提示することにより、ユーザビリティに関して、JIS T0601-1 が JIS T60601-1-6 を引用規格とし、更に JIS T60601-1-6 が JIS T62366-1:2022(2022年 2月発行) を引用するという、JIS 規格のみでユーザビリティ関連の引用規格を構成する形にしようとしています。
下記の WEB サイトでパブリックコメントの受付をしています。本サイトでは、上記 7件の JIS 規格原案の概要説明資料の他、規格原案の検討用に全文が掲載されたファイルを入手することができます。パブリックコメント募集期間は 9月 13日まで。案件番号は「495220093」となります。
参照 URL:
https://public-comment.e-gov.go.jp/servlet/Public?CLASSNAME=PCMMSTDETAIL&id=495220093&Mode=0
関連セミナー:
医療機器のサイバーセキュリティ関連規制、規格動向
| 2022年 6月の情報 |
欧州医療機器規制要求事項のタイムリーな遵守を確実にするための製造業者への通知について
2022年 6月 13日に、欧州委員会より、MDD 及び AIMDD の認証書を保有している製造業者に対するポジションペーパー(MDCG 2022-11)が発行されましたのでご紹介します。
解説:
MDR の適用日は 2021年 5月 26日ですが、MDD 及び AIMDD の認証書の有効期限の有効性は最長 2024年 5月 26日まで認められており、認証書の有効性が維持されている間は MDD 及び AIMDD の CE マーキングで機器を欧州に上市することを認める経過措置が実施されています。
この移行期間について、本ポジションペーパーは、MDRに基づく品質マネジメントシステムや技術文書の体制を準備するための期間をさらに確保することを意図しており、製造業者は、新規則の適用開始を延期するための猶予期間と受け止めるべきではない、と述べています。
現在までに 30 機関の Notified Body が MDR のもとで指定されており、MDD / AIMDD 認証書の約 80%を管理しています。多くの MDD / AIMDD 認証書が失効する直前となる 2024年 前半は、Notified Body がに全ての申請書類を評価することができない可能性があることが指摘されています。また、2022年 4月に行った Notified Body に対する調査では 75% の Notified Body が提出された 申請書 の 50% 以上が不完全であると判断したことを回答したということも述べています。
本ポジションペーパーは、最後に、早期に、完全かつ規制要件に適合した申請書類を提出し、余裕をもって MDR の認証書を取得することで、医療機器を上市し続けることが推奨されるという主旨を述べています。
参考:文書のダウンロード
MDCG 2022-11
MDCG Position Paper / Notice to manufacturers to ensure timely compliance with MDR requirements [PDF]
関連セミナー:
欧州医療機器規則(MD REGULATION)改正対策勉強会
欧州規制関連情報(スイス)
スイス国内の体外診断用医療機器規制の状況について
スイスは、IVDD の下で EU との MRA(Mutual Recognition Agreement)により EU 加盟国と同等と見なされてきましたが、IVDR については、その適用日(2022年 5月 26日)までに EU との間で MRA が締結されない場合は、IVDR の上ではスイスは「第三国」の扱いになるとされていました。
本件についての EU 委員会からのアナウンスは「スイスの間で MRA が、IVDR 適用日までに締結されなかった場合は」という記載であり、EU とスイスで MRA が締結されなかったことの発表はありませんが、テュフズード内部から得られる情報では「MRA は締結されていない」とのことです。
更にスイス連邦評議会は、この状況を受けて、国内の体外診断用医療機器を含む医療機器の供給を維持し、市場監視システムを維持するために、2022年 5月 4日に体外診断用医療機器に関する新規則(IvDO)を採択し、2022年 5月 26日付で発効しました。
解説:
IVD 製造業者の視点では、上記により IVDR 対策上、下記の解釈になる点に注意が必要となります。
その一方で、今後、スイスへの体外診断用医療機器の上市のためには以下が必要となります(一部移行措置期間あり)。
スイス体外診断用医療機器に関する新規則(IvDO)の発効に伴い、法規制文書は順次改訂が進められています。例えば、インシデント及び FSCA の報告に関する文書は、2022年 5月 26日より新しいバージョンに置き換わりました。体外診断用医療機器に関する臨床試験は、2022年 5月 26日より医療機器の臨床試験に関する規則(CTO-MedD)のもとで規制されます。
参照 URL:
EU 委員会: https://ec.europa.eu/health/latest-updates/notice-stakeholders-status-eu-switzerland-mutual-recognition-agreement-mra-vitro-diagnostic-medical-2022-05-24_en
Swissmedic: https://www.swissmedic.ch/swissmedic/en/home/news/mitteilungen/neue-regulierung-in-vitro-diagnostika.html
IvDO参考英訳: https://www.fedlex.admin.ch/eli/cc/2022/291/en
関連セミナー:
スイス体外診断用医療機器関連規制セミナー
国際規格関連情報 (ISO 10993-18:2020)
国際規格 ISO 10993-18:2020 / AMD1:2022 の発行 について
ISO 10993-18:2020 / AMD1:2022(ISO 10993-18:2020 追補1):不確かさの要因の決定 が、2022年 5月 11日付で発行されましたのでご紹介します。
解説:
国際規格 ISO 10993-18 は、医療機器に用いる材料についてのリスクマネジメントプロセスを用いた化学キャラクタリゼーションについて規定した規格です。その AMD1 / 追補1:版は、ISO 10993-18:2020 内の誤記修正、説明事項の差し替えが主な内容ですが、Annex E (Informative) Calculation and application of the analytical evaluation threshold(AET) の 「E.3 不確かさの要因の決定」については、内容が全面入れ替えとなっています。
Annex E を分析評価しきい値 (AET) の算出に際して参考にしている場合、AMD1 / 追補1:版の新しい記載内容をご参照ください。
参照 URL:
https://www.iso.org/standard/82241.html
関連セミナー:
ISO 10993 医療機器の生物学的安全性評価ビギナーズセミナー
欧州規格関連情報
MDR / IVDR に基づく UDI システムに関する質疑応答集の発行について
2022年 5月 20日に、欧州委員会より、MDR / IVDR に基づく UDI システムに関する質疑応答集(MDCG 2022-7)が発行されましたのでご紹介します。
解説:
MDR 第27条 及び IVDR 第24条により、医療機器へ唯一無二の UDI 及び Basic UDI-DI を割り当てることが義務付けられています。この質疑応答集には、UDI-DI、Basic UDI-DI、UDI に関するラベリング、システム及び処置パックなどの UDI ルール、キットと UDI のルールなどについての質問と、その回答が掲載されています。
掲載された質疑応答の要旨の例として、以下のものがあります。
Q: 単回使用機器の使用後、リプロセッシング業者がリプロセッシングした。新しい UDI-DI が必要か?
A: 必要となる。リプロセッシング業者が Manufacturer と見なされるため。
この他にも、例えば「Q: このような場合に新しい UDI-DI が必要となるか?」という主旨の質問とそれに対する回答が、具体事例と共に提示されています。
参照 URL:
https://ec.europa.eu/health/latest-updates/mdcg-2022-7-qa-unique-device-identification-system-under-regulation-eu-2017745-and-regulation-eu-2022-05-20_en
関連セミナー:
医療機器 UDI 規制とバーコード表示・ラベリングの留意点
欧州規制関連情報
IVDR に基づく安全性と性能のサマリーのテンプレートの発行について
2022年 5月 20日に、欧州委員会より、IVDR(欧州体外診断用医療機器規則)の安全性と性能のサマリー(summary of safety and performance: SSP)を作成するためのテンプレート(MDCG 2022-9)が発行されましたのでご紹介します。
解説:
IVDR において、製造業者は、Class C 及び Class D の機器について「安全性と性能のサマリー」を作成することが義務付けられています。安全性と性能のサマリーは、ノーティファイドボディによって審査され、EUDAMED を介して一般に公開されます。二種類のテンプレートが用意されており、一つは専門家によって検査を実施することを意図している機器のためのテンプレート、もう一つは自己検査(Self-testing)用機器のためのテンプレートです。
参照 URL:
MDCG 2022-9
https://ec.europa.eu/health/latest-updates/mdcg-2022-9-summary-safety-and-performance-template-2022-05-20_en
JIS 規格関連情報(パブリックコメント)
JIS T81001-1 パブリックコメント募集について
セキュリティに関する配慮が必要なヘルスソフトウェアに関する規格群である IEC 81001 シリーズに用いられる用語及び概念をまとめた国際規格 IEC 81001-1 の JIS 規格原案のパブリックコメントが、2022年 5月 26日に公示されましたのでご紹介します。
解説:
医療機器のソフトウェアを含むヘルスソフトウェアのサイバーセキュリティについては、 2021年に IEC 81001-5-1:2021 という国際規格が発行されており、今後、この IEC 81001-5-1 が各国の医療機器法規制に取り入れられて用いられるものと予測されています。
IEC 81001-5-1 に基づいた JIS 規格は現在まだ準備中ですが、IEC 81001-5-1 に先立ち、IEC 81001-5-1 を含む規格群 IEC 81001 シリーズに用いられる用語及び概念をまとめた国際規格 IEC 81001-1 の JIS 規格版である「JIS T81001-1 ヘルスソフトウェア及びヘルス IT システムの安全、有効性及びセキュリティ- 第1部:原則及び概念」の原案がパブリックコメントに出され、パブリックコメントのサイトで閲覧できるようになりました。IEC 81001-5-1 の検討の上で理解が必要な規格となりますので、対象となる医療機器のメーカーの皆様には早めの検討をご推奨します。
下記の WEB サイトでパブリックコメントの受付をしています。本サイトでは、本 JIS 規格原案の概要説明資料の他、規格原案の検討用に全文が掲載されたファイルを入手することができます。パブリックコメント募集期間は 7月 25日まで。案件番号は「495220040」となります。
参照 URL:
https://public-comment.e-gov.go.jp/servlet/Public?CLASSNAME=PCMMSTDETAIL&id=495220040&Mode=0
関連セミナー:
医療機器のサイバーセキュリティ関連規制、規格動向
| 2022年 5月の情報 |
MDR 欧州整合規格リスト小改正(Amendment)について
2022年 5月 17日(OJ は 2022年 5月 11日付)、欧州医療機器規則(MDR)の整合規格リストの Amendment が発行されましたのでご紹介します。
解説:
今回の Amendment によりリスクマネジメント規格 EN ISO 14971:2019 / A11:2021 がMDR 欧州整合規格となった点が注目されます。
参照 URL:
https://eur-lex.europa.eu/eli/dec_impl/2022/757/oj
関連セミナー:
欧州規格版 ISO 14971・EN ISO 14971:2019 / A11:2021 セミナー
欧州規制関連情報
IVDR 110条(3)の下での移行措置期間中の Significant Change の解釈に関わるガイダンス文書発行について
2022年5月、MDCG(医療機器調整グループ)は、IVDD の認証を取得して CEマーキングを表示した体外診断用医療機器についての、IVDR 適用日以降の Significant Change の解釈についてガイダンスとして取りまとめ、MDCG 2022-6として発行しましたのでご紹介いたします。
解説:
本ガイダンス文書 MDCG 2022-6 は、IVDR 110条(3)の「設計及び意図された目的の Significant Change」の概念を明確にすることを目的に作成されました。IVDD の認証を取得して CEマーキングを表示した体外診断用医療機器のうち、IVDR 適用日以降に引き続き EU 市場に Place on the market を継続する機器において変更事項が発生した場合、それが「設計及び意図された目的の Significant Change」に該当するか否かを判断するのに役立つ内容となっています。「設計及び意図された目的の Significant Change」の基本的な解釈の記載、Significant Change に該当する事例などの他、判断のためのデシジョンツリーも掲載されています。
なお「設計及び意図された目的の Significant Change」に該当する変更を実施した場合、IVDD の認証に基づいた CE マーキングでは EU 市場への Place on the market を継続することができなくなり、IVDR の CE マーキングへの切り替えが必須になる点に注意が必要です。
参考URL:
MDCG 2022-6
Guidance on significant changes regarding the transitional provision under Article 110(3) of the IVDR [PDF}
関連セミナー:
欧州 IVD 規則 (IVD Regulation) 改正対策勉強会
欧州規制関連情報
IVDR 欧州整合規格リスト小改正(Amendment)について
2022年 5月 11日付で、欧州体外診断用医療機器規則(IVDR)の整合規格リストの Amendment が発行されましたのでご紹介します。
解説:
今回の Amendment によりリスクマネジメント規格 EN ISO 14971:2019 / A11:2021 が IVDR 欧州整合規格となった点が注目されます。
参照 URL:
https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32022D0729&from=EN
関連セミナー:
欧州規格版 ISO 14971・EN ISO 14971:2019 / A11:2021 セミナー
欧州規制関連情報
医療機器と医薬品の境界線に関するガイダンス文書の発行について
欧州委員会より、欧州医療機器規則(MDR)に基づく医療機器と医薬品の境界線に関するガイダンス文書(MDCG 2022-5)が発行されましたのでご紹介します。
解説:
医療機器に関する欧州規則(EU)2017/745(MDR)と医薬品に関する欧州指令 2001/83/EC の境界の明確化は、法規制を適切に実施し正しく解釈および施行するために重要となります。
医療機器の性質も医薬品の性質もあわせ持つ製品に対して、これらの二つの法的枠組みの境界を確立するためのいくつかの規定が定められています。その中で本ガイダンス文書は、欧州全体での MDR の均一な適用を支援するために、これらの規定を明確にする詳細な説明と例を提供しています。例えば、医薬品を不可欠な部分として補助的に組み込んでいるが、法的には「医療機器」と見なされる例として、ヘパリンまたは抗生物質でコーティングされたカテーテル、抗生物質を含む骨セメントなどが挙げられています。
参考:文書のダウンロード
MDCG 202 –5
Guidance on borderline between medical devices and medicinal products under Regulation (EU) 2017/745 on medical devices [PDF]
関連セミナー:
欧州医療機器規則(MD REGULATION)改正対策勉強会
規制遵守責任者のための MDR/IVDR 勉強会
MDSAP 関連情報
改訂版 MDSAP Audit Approach の発行について
2022年 4月 15日付で、米国 FDA(食品医薬品局)より改訂版 MDSAP Audit Approach (MDSAP AU P0002.007)が発行されましたのでご紹介します。
解説:
MDSAP Audit Approach 改訂 007 版では、いくつかの情報の更新がなされました。
2021年 9月から有効となった不適合の格付け(MDSAP AU 0037.001)が参照できるようになった他に、附属書6 が新設され、組織の認証スコープから受容できる除外事項について、国別にまとめられました。
また、オーストラリアのTGA 1989 及びTG(MD)R 2002 の規制条項番号の更新、ブラジル GMP が RDC 16/2013 から RDC ANVISA 665/2022 に代わったことに伴う条項番号の更新、日本の QMS 省令の条項番号の更新も行われています。ブラジル GMP は、国内法規制の標準ルールにのっとって条項番号を採番したことによるものとなっています。
参照 URL:
https://www.fda.gov/media/157947/download
関連セミナー:
MDSAP AUDIT APPROACH 文書解説セミナー
| 2022年 4月の情報 |
欧州規格関連情報
EN ISO 14971:2019/A11:2021の発行について
2021年 12月に CEN/CENELEC より、EN ISO 14971:2019/A11:2021 が発行されましたのでご紹介します。
解説:
EN ISO 14971:2019/A11:2021 の要求事項は国際規格版 ISO 14971:2019と同じものとなります。ただし、EN ISO 14971:2019/A11:2021 には ISO 14971:2019 にはない附属書 ZA 及び ZB が追加されており、それぞれ MDR 及び IVDR の附属書 I 安全性と性能の要求事項への適合を示す上で 考慮すべき点が記載されています。医療機器製造業者は、この附属書ZA 及び ZB を参考にして MDR/IVDR の要件を満たすために必要なリスクマネジメント活動を検討する必要があると考えられます。
EN ISO 14971:2019/A11:2021 が欧州整合規格として欧州官報に公示された後、附属書ZA 及び ZB を含めて EN ISO 14971:2019/A11:2021 への対応をすることにより、MDR/IVDR の附属書 I 安全性と性能の要求事項へ適合しているとみなされることとなります。
補足:2022年 5月 11日付で、欧州体外診断用医療機器規則(IVDR)の欧州整合規格リストの Amendment が発行されました。その Amendment によりリスクマネジメント規格 EN ISO 14971:2019 / A11:2021 が IVDR 欧州整合規格となっています。
※ 本規格 EN ISO 14971:2019/A11:2021 と、国際規格版 ISO 14971:2019 の違いは何か?を解説するセミナーを開催します。
参照 URL:
https://standards.cencenelec.eu/dyn/www/f?p=305:110:562161845733001::::FSP_ORG_ID,FSP_PROJECT,FSP_LANG_ID:581003,75110,25&cs=19A657771114E2C502DE825EE67EDCA26
関連セミナー
欧州規格版 ISO 14971・EN ISO 14971:2019/A11:2021 セミナー
厚生労働省関連情報
医療機器プログラムの該当性ガイドラインについて
医療機器プログラムへの該当性についてのガイドライン(「プログラムの医療機器該当性に関するガイドラインについて」薬生機審発 0331 第1号、薬生監麻発 0331 第15号)及びプログラム医療機器事例データベース が、2022(令和4)年 3月 31日付で公表されましたのでご紹介します。
解説:
プログラムの医療機器への該当性に関する基本的考え方についての通知が、平成 26年 11月 14日付通知としてで発出されました。その後、汎用 PCや携帯情報端末等にインストールして使用するプログラム開発が進んだこと、また諸外国におけるプログラム医療機器の該当性及びクラス分類に関するガイダンスが発出されたことをきっかけに、日本国内におけるプログラムの医療機器該当性をより明確にし、プログラム開発において事業の予見可能性を高めることを目的として本通知が発出されました。また併せて、プログラム医療機器事例データベースが公開されています。これは、開発するプログラムの医療機器への該当性を判断する一助となるものです。
参考URL:
https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000179749_00004.html
参考:文書のダウンロード
薬生機審発0331第1号、薬生監麻発0331第1号
令和4年3月31日
プログラムの医療機器該当性に関するガイドラインについて [PDF]
医療機器プログラム事例データベース
厚生労働省関連情報
医療機器プログラムの設計のみを行う製造所における責任技術者の取扱いに係わる通知について
2022年 3月 30日に、厚生労働省より「医療機器プログラムの設計のみを行う製造所における責任技術者の取扱いについて(薬生機審発 0330第1号 令和4年 3月 30日)」が発出されましたのでご紹介します。
解説:
医療機器プログラムの設計を行う製造所においては、製造所登録が必要とされてきました。令和3年12月22日に内閣府の規制改革推進会議が公表した「当面の規制改革の実施事項」により、医療機器プログラムの設計のみを行う製造所において、プログラムの開発者は必ずしも製造業の登録を受けた所在地で勤務する必要はないとの見解が明示されました。
参考:文書のダウンロード
薬生機審発 0330第1号
令和4年 3月 30日
医療機器プログラムの設計のみを行う製造所における責任技術者の取扱いについて [PDF]
関連セミナー:
QMS 省令要求事項解説 - 令和3年 厚生労働省令 第60号改正版
厚生労働省関連情報
医療機器及び体外診断用医薬品の製造管理及び品質管理の基準等に係る質疑応答集の発行について
2022年 3月 4日に、厚生労働省より「医療機器及び体外診断用医薬品の製造管理及び品質管理の基準等に係る質疑応答集について(その4)(事務連絡 令和4年3月4日)」が発出されましたのでご紹介します。
事務連絡
令和4年3月4日
医療機器及び体外診断用医薬品の製造管理及び品質管理の基準等に係る質疑応答集について(その4)
解説:
QMS 省令改正(令和 3年厚生労働省令第60号改正版)に伴う基準適合証の考えなどが明示されています。例えば、改正QMS省令の施行日(2021年3月26日)より以前に実施された、旧QMS省令に基づく適合性調査によって交付された基準適合証の有効性に対する厚生労働省の見解が記載されています。
参考:文書のダウンロード
事務連絡
令和4年3月4日
医療機器及び体外診断用医薬品の製造管理及び品質管理の基準等に係る質疑応答集について(その4)[PDF]
関連セミナー
QMS 省令要求事項解説 - 令和3年 厚生労働省令 第60号改正版
カナダ関連情報
「List of Medical Devices - Notification of Shortages」の発行及びカナダ医療機器規則改正について
2022年 3月 2日に、カナダで「List of Medical Devices - Notification of Shortages」が発行されましたのでご紹介します。また、このリスト発行に先立ち、カナダ医療機器規則も改正されています(SOR/2021-199)。
解説:
本リスト(List of Medical Devices - Notification of Shortages)に掲載された種類の医療機器は、カナダで市場の要求に応じた供給ができないか又はその恐れがある場合は、製造業者は速やかにカナダ保健省に通知しなければならないという制度が新設されていますのでご注意ください。
カナダ保健省は「COVID-19 対応のための対策」と説明しており、例えば、医療用マスク、外科用グローブ、赤外線式体温計、パルスオキシメーター、ECMO などの医療機器がこのリストに含まれています。
参照 URL:
https://www.canada.ca/en/health-canada/services/drugs-health-products/medical-devices/shortages/mandatory-reporting.html
関連セミナー:
カナダ医療機器法規制セミナー
| 2022年 3月の情報 |
欧州規制関連情報
欧州医療機器規則(MDR)の安全性と臨床性能のサマリー(SSCP)に関する改訂版ガイダンス文書の発行について
2022年 3月 24日に、欧州委員会より、欧州医療機器規則(MDR)の安全性と臨床性能のサマリー(SSCP)に関する改訂版ガイダンス文書(MDCG 2019-9 - Rev.1)が発行されましたのでご紹介します。
解説:
MDR の第32条により、Class III 医療機器と埋込み医療機器の製造業者は、安全性と臨床性能のサマリー(SSCP)を作成することが義務付けられています。その SSCP はノーティファイドボディにより審査され、EUDAMEDを介して公表されます。2019年 9月 26日発行の初版 MDCG 2019-9 から改訂された主な記載は、SSCP と EUDAMED の Basic UDI-DI との関連性です。例えば、複数の機器から構成されるシステム機器の場合、システム機器に対する Basic UDI は、SSCP 内の一節「Basic UDI」に記載し、SSCP の EUDAMED 登録時にも使用します。システム機器に含まれる機器それぞれの Basic UDI は、SSCP内の一節「機器の説明」に記載すると明記されています。
参照 URL:
https://ec.europa.eu/health/latest-updates/update-mdcg-2019-9-rev1-summary-safety-and-clinical-performance-2022-03-24_en
関連セミナー:
欧州医療機器規則 MDR 最新情報と医療機器の臨床評価
欧州医療機器規則(MD REGULATION)改正対策勉強会
医療機器 UDI 規制とバーコード表示・ラベリングの留意点
国際規格関連情報
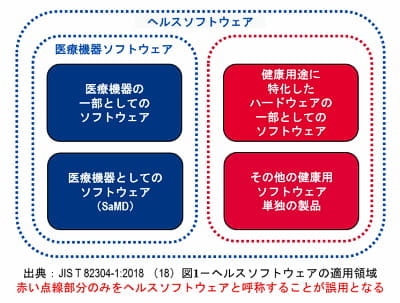
「ヘルスソフトウェア」という用語の意味について
例えば、IEC 81001-5-1:2021 は「ヘルスソフトウェア」を対象にした国際規格として制定されています(2月情報欄参照)。このヘルスソフトウェアという用語は誤用されることがあり、誤解を招き、本件に関連する JIS 規格を議論する会議においても注意喚起がありましたので「医療機器ソフトウェア」との関連性を整理して解説し情報を共有いたします。
解説:
まず「医療機器の一部としてのソフトウェア」と「医療機器としてのソフトウェア(SaMD)」を足した概念を「医療機器ソフトウェア」と言います。なお「医療機器としてのソフトウェア(SaMD)」とは単体のソフトウェア製品で、医療機器としての意図した使用目的(例:診断)を持つものを指します。
次に「ヘルスソフトウェア」の定義は「個人の健康を管理、維持若しくは改善するために使用することを意図するソフトウェア、ケアを提供するために使用することを意図するソフトウェア」と「医療機器ソフトウェア」を足し合わせたものとなります(図1 参照)。
ただし、その一方で「ヘルスソフトウェア」という用語は「健康用途に用いられるソフトウェアだが医療機器の規制対象とならないもの(即ち医療機器ソフトウェアを除外したもの)」を指すという誤った用法で使われるケースがあり、医療現場等での混乱の原因になるとのことです。「ヘルスソフトウェア」という用語を「規制対象にならない健康関連ソフトウェア」という意味で用いることは国際的な用語の定義に合わないのでご注意いただきたいとのことです。
欧州規制関連情報
MDR 第120条 の移行措置規定についての適切なサーベイランス審査に関するガイダンス文書の発行
2022年 2月 16日に、欧州委員会より、MDR第120条 の移行措置規定についての適切なサーベイランス審査に関するガイダンス文書(MDCG 2022-4)が発行されましたのでご紹介します。
解説:
このガイダンス文書(MDCG 2022-4)は、ノーティファイドボディが欧州医療機器規則(MDR)第120条に沿って実行しなければならない監査活動の詳細を説明しています。欧州医療機器指令(MDD)に基づいて CE マーキングを表示した機器が Place on the market されている場合、ノーティファイドボディはサーベイランス監査を継続しなければならないのは当然のことですが、非通知監査もその監査活動の一部であり続けると明記されています。そして、定期的安全更新報告(PSUR)に代表される MDR で規定されている製造業者の市販後監視活動は、その監査対象であると言及されています。また、この監査活動にはサンプリングベースの技術文書評価が含まれており、移行期間の終了までの期間、または EC 認証書の有効期限が 2024年 5月 26日より前の場合は発行された EC 認証書の有効期間が終了するまでの期間、MDD 又は AIMDD に準拠した技術文書評価が実行されます。
参照 URL:
https://ec.europa.eu/health/latest-updates/mdcg-2022-4-guidance-appropriate-surveillance-regarding-transitional-provisions-under-article-120-2022-02-16_en?utm_source=CleverReach&utm_medium=
関連セミナー:
欧州医療機器規則(MD REGULATION)改正対策勉強会
規制遵守責任者のための MDR/IVDR 勉強会
欧州規制関連情報
スイス単一登録番号(CHRN)に関する情報シートの発行
2022年3月1日に、Swissmedic(スイス医薬品局)より、スイス単一登録番号(Swiss Single Registration Number: CHRN)に関する改訂版情報シートが発行されましたのでご紹介します。
解説:
CHRNは、Swissmedic がスイスのエコノミックオペレータ(製造業者、指定代理人、輸入業者等)へ、要求に応じて割り当てる単一識別番号です。スイス連邦と EU の間で医療機器適合性評価の相互承認に関する運用上の合意がない場合、エコノミックオペレータと医療機器固有識別(UDI)の登録は Swissmedic によって実行されます。現在、Swissmedic はこれらを登録するための新しいデータベースを設計しています。スイス連邦と EU の規制の同等性を継続的に目指しているため、このデータベースは EUDAMED と同様になるとしており、一括アップロードでの医療機器登録も可能になるとしています。
参照 URL:
https://www.swissmedic.ch/swissmedic/en/home/medical-devices/medizinprodukte-datenbank.html
関連セミナー:
欧州医療機器規則(MD REGULATION)改正対策勉強会
規制遵守責任者のための MDR/IVDR 勉強会
米国規制関連情報
QSR 改正案のパブコメ募集について
2022年 2月 23日に、米国 FDA(食品医薬品局)より、医療機器 QSR(Quality System Regulation)の改正に係わる提言案が発行されましたのでご紹介します。
解説:
QSR とは、米国における総合的な医療機器品質システムのための要求事項で、連邦規則 21 CFR Part 820 を指します。20年以上前に本規則が施行されて以来、これまで十分かつ効果的な要件を提供してきましたが、品質システムに対する法規制の期待は進化し続けています。そこで、FDAは、FDA 法規制下にある医療機器について、品質管理システム(QMS)に関する現在の国際認知規格 ISO 13485:2016 を明示的に要求することを提案しました。
この提言がまとまった場合の移行期間や、ISO 13485:2016 と QSR との間で異なる用語及び定義など、現時点で不明な点については、引き続き情報収集が必要と思われます。
パブコメ(意見公募)の締め切りは、2022年 5月 24日とされています。
参照 URL:
https://www.federalregister.gov/documents/2022/02/23/2022-03227/medical-devices-quality-system-regulation-amendments
関連セミナー:
FDA 規制 & QSR 勉強会
MDSAP AUDIT APPROACH 文書解説セミナー
厚生労働省関連情報
登録認証機関等に対する立入検査の実施要領の一部改正について
令和 4年(2022年)2月 22日に、厚生労働省から、通知「登録認証機関等に対する立入検査の実施要領の一部改正について」が発出されましたのでご紹介いたします。
解説:
本通知には、行政当局による登録認証機関の立入検査と立会検査の手順が記載されていますが、注目点は登録認証機関への立入検査だけでなく立会検査(登録認証機関が実施する調査への行政当局による立会い)についての記述もあることです。
立会検査において、立会職員は「登録認証機関が実施する調査対象施設に対する適合性調査等業務に影響を及ぼさない」ことが原則ですが「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律施行規則(昭和36 年厚生省令第1号)第137 条に規定する事実があった場合においてはこの限りではない」とされています。
参考:文書のダウンロード
薬生機審発0222 第2号
令和 4年 2月 22日
登録認証機関等に対する立入検査の実施要領の一部改正について [PDF]
| 2022年 2月の情報 |
国際規格関連情報
国際規格 IEC 81001-5-1:2021 の発行について
2021年12月に、国際電気標準会議(IEC)より、IEC 81001-5-1:2021 が発行されましたのでご紹介します。
解説:
本規格は、このWebページの2021年 9月でご紹介した通り、ISO 13485 及び IEC 62304 に基づいて、ソフトウェアを持つ医療機器(ソフトウェア自体が医療機器である場合を含む)の開発を進める上で必要となる、ソフトウェアライフサイクルを通した「ITセキュリティ」に関する取組みを規定した規格です(ただし、ISO 13485、IEC 62304 を引用規格とはしていません)。
注目すべき点の一つは、この規格がヘルスソフトウェアの製造業者をターゲットにしていることです。すなわち、その適用範囲が、医療機器だけではなく、ヘルス分野で使用されている他のソフトウェア、例えば、フィットネスアプリ、介護サービス計画支援アプリも含まれています。
参照 URL:
https://www.iso.org/standard/76097.html
関連セミナー:
医療機器のサイバーセキュリティ関連規制、規格動向
厚生労働省関連情報
医療機器のサイバーセキュリティの確保及び徹底に係る手引書について
令和 3年(2021年)12月24日に、厚生労働省から、通知「医療機器のサイバーセキュリティの確保及び徹底に係る手引書について」が発出されましたのでご紹介いたします。
解説:
この手引書は、国立研究開発法人日本医療研究開発機構医薬品等規制調和・評価研究事業「医療機関における医療機器のサイバーセキュリティに係る課題抽出等に関する研究」及び一般社団法人日本医療機器産業連合会が、国際医療機器規制当局フォーラム(IMDRF)のガイダンス文書「Principles and Practices for Medical Device Cybersecurity」の内容を基に作成し、厚生労働省が認知したものです。IMDRF ガイダンスの要求事項を踏まえつつ、医療機器製造販売業者等のもつべき責任など、日本の状況に応じた解釈を加筆した内容となっています。
参考:文書のダウンロード:
薬生機審発 1224 第1号 / 薬生安発 1224 第1号
令和 3年 12 月 24 日
医療機器のサイバーセキュリティの確保及び徹底に係る手引書について [pdf]
関連セミナー:
医療機器のサイバーセキュリティ関連規制、規格動向
欧州規制関連情報
IVDR に基づいた体外診断用医療機器の臨床的証拠(Clinical Evidence)の一般原則に関するガイダンス文書について。
2022年 1月27日に、欧州委員会より、体外診断用医療機器の臨床的証拠の一般原則に関するガイダンス文書(MDCG 2022-2)が発行されましたのでご紹介します。
解説:
十分な臨床的証拠(Clinical Evidence)を持つことは、欧州体外診断用医療機器規則(IVDR)の安全と性能の要求事項(GSPR)への適合性を示す上での必須事項となります。このガイダンス文書は、性能評価プロセスの原則、リスク管理と性能評価プロセスの相互関係、および市販後調査(PMS)と市販後性能フォローアップ(PMPF)までの個々のステップの詳細な説明など、臨床的証拠(Clinical Evidence)の重要な側面をカバーしています。
27ページ目には、質問集が掲載されていますが、製造業者が IVDR の適合性評価手順に臨むための準備で参考となるものです。
※ 欧州 IVD 規則 (IVD Regulation) アドバンスド・セミナーで本ガイダンスの解説を予定しています。
参考:文書のダウンロード
MDCG 2022-2 Guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices (IVDs) [PDF]
関連セミナー:
欧州 IVD 規則(IVD Regulation)アドバンスド・セミナー
欧州規制関連情報
欧州体外診断用医療機器規則(IVDR)改正版について。
欧州体外診断用医療機器規則(IVDR)の移行期間延長が反映された改正版が EU 官報に掲載されましたのでご紹介します。
解説:
具体的な改正内容は、当初提案通りとなります。「2021年 11月の情報」 の 「欧州規制関連情報・IVDR の改正提案について」 という記事で解説しています。また「2021年 12月の情報」 の 「IVDR の移行処置規定改正について」 という記事で、欧州委員会によるプレスリリースの和訳をご提供しています。
https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CONSIL%3APE_79_2021_REV_1&qid=1643182084832
関連セミナー:
欧州 IVD 規則(IVD Regulation)改正対策勉強会
| 2022年 1月の情報 |
欧州規制関連情報
MDR 及び IVDR の欧州整合規格リスト・初版の修正について。
欧州委員会が、2022年 1月 4日及び 6日の欧州官報で、欧州医療機器規則(MDR)及び欧州 IVD 医療機器規則(IVDR)の 欧州整合規格リストの修正版を公示しましたのでご紹介します。
解説:
今回発行された MDR / IVDR 欧州整合規格リストは、2021年 7月に発行された初版のリストに対するアップデートを示したものとなります。CEN(欧州標準化委員会)及び CENLEC(欧州電気標準化委員会)が最新の技術的・科学的進歩を考慮し、各規則の要求事項に適合するために発行した規格の中から、新たに MDR の欧州整合規格として 9つの EN 規格、IVDR の欧州整合規格として 5つの EN 規格を参照しています。
ただし、欧州整合規格として掲載された規格の数はまだ限定的であり、今後も順次追加する必要があるものと考えられます。
MDR / IVDR に共通して規定された 4つの規格のうち EN ISO 13485:2016 は、2021年9月に発行された EN ISO 13485:2016+A11:2021 を含めて整合規格の対象となります(EN ISO 13485:20116+A11:2021 については、2021年9月の情報を参照ください)。
それ以外の、両規則に共通して規定された規格として、表示物に用いるシンボル規格(EN ISO 15223-1:2021)、滅菌関連の2つの規格(EN ISO 11737-1:2018, EN ISO 13408-6:2021)があります。
参照 URL:
MDR: https://eur-lex.europa.eu/eli/dec_impl/2022/6/oj#ntr5-L_2022001EN.01001101-E0005
IVDR: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32022D0015&qid=1641862665368
参考:文書のダウンロード:
MDR: Official Journal of the European Union (4 January 2022) [PDF]
IVDR: Official Journal of the European Union (6 January 2022) [PDF]
Site Selector
Global
Americas
Asia
Europe
Middle East and Africa