
医療機器市場における承認に確信を
医療機器市場における承認に確信を
テュフズードは、欧州連合(EU)のMDR(Regulation (EU) 2017/745)のノーティファイドボディとして認定された世界初の組織の一つです。
2つのEUノーティファイドボディと、世界の30か所以上、750名以上の医療機器のエキスパートを擁する当社は、欧州MDRの下でグローバルに認証サービスを提供できる最大のEUノーティファイドボディの一つです。
MDR認証プロセスについて、下記のフォームまたはこちらよりお問い合わせください。
MDRサービス依頼はこちら
製造業者は、ノーティファイドボディによる適切な適合性評価手順を、製品のクラス分類に基づいて申請する必要があります。テュフズードのMDRサービスの詳細については、テュフズードノーティファイドボディのMDR申請手続きに関する各ウェブページをご覧ください。
また、EUのMDRに関する詳しい情報については、FAQページをご参照ください。
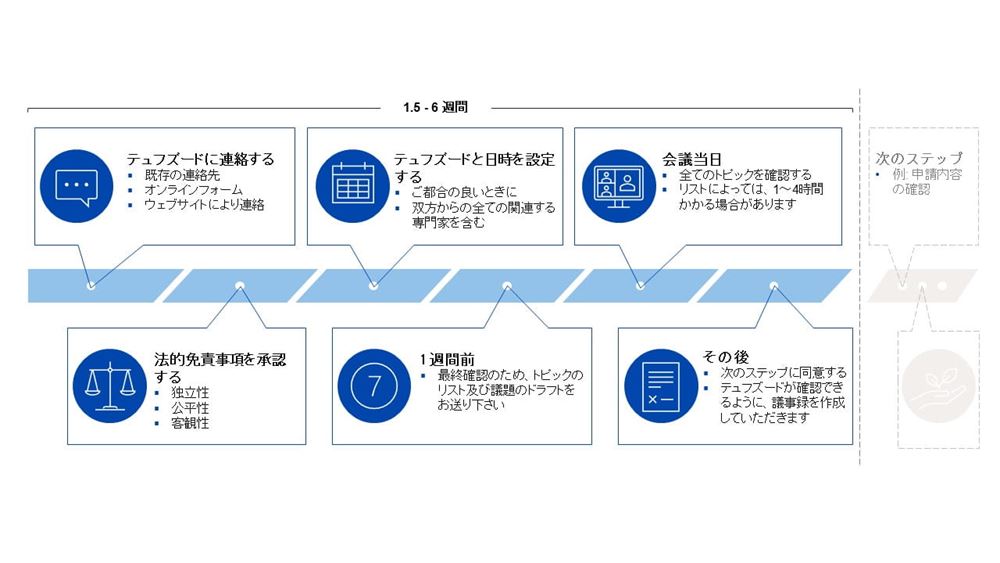
テュフズードに正式なMDR申請を提出する前に構造化された対話を行う目的は、申請プロセスとフォーム及び提出書類のタイミング、手続き、規制の側面を明確にすることです。
構造化された対話は、適合性評価の申請前のお客様との会議に限定され、評価とは無関係です。従って、弊社の独立性、公平性、客観性を最大限示すために、構造化された対話サービスはMDRフレームワーク合意書とは独立して発注されるものとします。
以下のタブを展開すると、可能なトピック、プロセス、及び構造化された対話の申請方法を確認できます。
プレ申請及び申請プロセス
製造業者の責務
適合性評価手順
テュフズードノーティファイドボディの既存のお客様:
テュフズードへの申請を検討中のお客様:
欧州医療機器規則(MDR)2017/745は、2017年4月に欧州連合官報に掲載され、すべてのEU加盟国における規制審査および承認プロセスを調和させるために策定されました。1990年代にEUの初期枠組みが実施されて以来、医療機器技術における大きな進歩を反映させています。
MDRの要求事項は、2021年5月26日付で、EU域内で販売されるすべての医療機器に適用されるようになりました。しかしMDRの特定の要件の実施は、欧州医療機器指令(93/42/EEC(MDD))および能動埋込型医療機器指令(90/385/EEC(AIMDD)の下で以前に承認された医療機器に対しては、最長で2028年12月まで延長されます。
2023年3月15日に、EU規則2023/607が正式に制定され、医療機器の製造業者には医療機器認証を行う時間がより長く与えられ、医療機器が不足するリスクが軽減されました。改正された規則では、MDD/AIMDDの下で既に市販されている製品について、MDRの下での新しい規則に適応するために、より長い移行期間が導入されました。
新たなMDR移行タイムラインの主要要素に関する詳しい情報については、MDR FAQで参照できます。
テュフズードは引き続き、新たな期限にもかかわらず、製造業者には、すぐに行動することを強く奨励します。EU MDRの手順は複雑であることが知られており、時にはより長い処理時間を必要とします。テュフズードは、早い段階から順調にキャパシティを増強し、製造業者と常に緊密なやり取りを行っています。しかし、製造業者は移行タイムラインの終盤での遅延を避けるために、新しい状況に積極的に取り組み、計画に全速力で取り組む必要があります。
EUは世界最大の医療機器市場の一つです。2020年時点でEUの市民は7億4,764万人で、米国の人口の3億3,300万人の2倍です。人工知能(AI)や遠隔監視機能などの技術的進歩もあり、EUの医療機器の製造業者の売上高は、2027年には1,700億ユーロを超えると予想されています。したがって、EUは、大手企業から革新的な新興企業まで、すべての医療機器製造業者にとって重要な市場です。
MDRの最も重要な要件は次のとおりです。
MDRでは、MDDおよびAIMDDの対象外であった製品を含む、広範な医療機器に適用されます。新たにカバーされる医療機器の具体的な例としては、カラーコンタクトレンズや美容インプラント機器などの医療目的を持たない機器が挙げられます。また、病気やその他の健康状態の「予見および予後」を目的として設計された機器も対象に含まれます。
機器の製造業者は、新しい規則の遵守について最終的に全ての責任を負う人物を、組織内で最低1名任命する必要があります。また、組織はその人物の特定の資格について、要求される業務に応じて文書化する必要があります。中小企業やスタートアップ企業にはいくつかの特別な救済措置が適用される場合があります。
MDRの附属書VIIIには、医療機器のクラス分類を規定する要件が詳述されています。いくつかの例では、MDR分類の要件はMDDまたはAIMDDの要件よりも厳格であり、その結果、いくつかの機器に対してより高いリスククラスが割り当てられ、従来よりも厳しい要件を満たす必要があります。
医療機器の製造業者は、医療機器がどのリスククラスにあるかにかかわらず、安全性、性能、さらに新しい要件として医療機器の利点を裏付ける十分な臨床的エビデンスを提供することが常に求められています。他の情報源から十分な臨床データが入手できない場合、臨床試験は依然として必須であり、これも医療機器のリスククラスに依存しません(MDR61条)。すべての機器について、製造業者はMDR Annex XIV Part Aに記載されている臨床評価計画を持つものとし、そこで製造業者は安全性、性能、および利益について期待される結果パラメータを提供するものとします。
埋め込み機器やクラスIII機器などの特定の医療機器には、臨床調査の必要性に関する独自のMDR規則があります(MDR 61条4.)。技術文書へのアクセスを確保するために、評価対象の機器の製造業者と同等の機器の製造業者との間の契約上の合意が必要になった(MDR, 61条5.)ため、これらのクラスIIIおよび埋め込み機器に対して広く使用されている同等性アプローチはより厳格になりました(MDR, Annex XIV Part A)。埋め込み機器およびクラスIII機器の場合、製造業者は、医療機器の安全性、性能、および利点に関する詳細な臨床データを含む、医療機器の専門ユーザーが理解できるだけでなく、該当する場合は患者も理解できる公開文書、いわゆる安全性と臨床性能の要約(Summary of Safety and Clinical Performance, SSCP)を毎年提供する必要があります。この文書は、EUDAMEDデータベースで公開されます。SSCPは、製造業者が遵守しなければならない専用の市販後調査プロセスの一部です(MDR 32条)。臨床データに特化した市販後調査プロセスは、MDR Annex XIV Part Bの市販後臨床フォローアップで詳細に管理されています。
クラスIII埋め込み機器およびルール12に従ったクラスIIb機器については、臨床評価コンサルテーション手順(Clinical Evaluation Consultation Procedure, CECP)という新しいコンサルテーションプロセスが開始されました。欧州委員会の独立した専門家委員会は、ノーティファイドボディの臨床評価審査報告書を審査し、臨床データ評価の結果について追加の科学的意見を提供します(MDR 54条)。
製造業者は、臨床試験が必要か否かを決定する際に、同等性のエビデンスの使用に関するMDRの厳格な要件を慎重に検討しなければなりません。
MDRでは、医療機器に機器固有識別子(UDI)方式の使用を義務付けています。本要求事項により、製造業者および関係当局がサプライチェーンを通じて特定の機器を追跡する能力が向上し、安全リスクが発覚した医療機器の迅速かつ効率的なリコールを支援することが意図されています。また、欧州医療機器データバンク(Eudamed)は、承認された医療機器に関する情報への効率的なアクセスを提供するために拡張されました。
ノーティファイドボディは、機器の再利用、特に洗浄、消毒、滅菌、保守、機能試験と使用または再処理に関する取扱説明書に関する側面について、クラスI再利用可能な手術器具の適合性評価に関与する必要があります。
MDRは、ノーティファイドボディによる市販後監視(PMS)の権限の強化を義務付けています。非通知審査は、製品サンプルのチェックや製品テストとともにEUの施行体制を強化し、安全ではない機器によるリスクを軽減するために役立ちます。多くの場合、製造業者には、毎年、安全性と性能の報告書が求められます。
医療機器の開発プロセスが複雑なことと、欧州MDRに盛り込まれた厳格な要件を考え合わせると、ほぼ全ての機器の製造業者にとって移行プロセスが複雑かつ時間のかかるものとなるでしょう。さらに、これまでにMDDまたはAIMDDの下で認証を受けている機器の製造業者も、MDRの要件からは免除されていません。引き続き販売される機器は、MDRの規定に従って、再度認証を受ける必要があります。
さらに、EUノーティファイドボディは、クラスI機器[1]を除き、MDRの範囲内にあるすべての医療機器の承認及び認証に関与しなければなりません。多数の医療機器に対してノーティファイドボディによる審査および承認が必要となるため、プロセスの進行に遅延が予想されます。したがって、医療機器の製造業者に対しては、適時かつ効率的なMDRの審査及び認証を達成に向け、必要なステップを計画するために、製品開発プロセスの早い段階でノーティファイドボディと協議することをおすすめします。事前準備と迅速な行動が鍵となります。
[1] 無菌状態で市場に出回っているクラスIの機器、計測機能を備えている機器、または再利用可能な手術器具は、ノーティファイドボディの関与が必要です。




IVD機器向けのEMC試験個別規格の最新版「IEC 61326-2-6:2020」が発行され、試験の考え方が旧規格より大幅に変更されました。
もっと知る


国内薬機法において、すべての能動型医療機器は2023年3月より「JIS T 0601-1-2:2018(Ed.4.0)」に適合する必要があります。
もっと知る

Site Selector
Global
Americas
Asia
Europe
Middle East and Africa