
体外診断用医療機器のEU市場アクセスを達成
体外診断用医療機器のEU市場アクセスを達成
体外診断用医薬品規則(IVDR)は、体外診断用医療機器をEU市場に上市し、利用を可能にし、使用開始するための現行の欧州連合(EU)の法規制です。
IVDR規則は2017年5月5日に欧州官報に掲載され、2017年5月26日に発効し、体外診断用医療機器に関するEUの旧指令(98/79/EC)を段階的に置き換えてきました。
EU規則であるため、EU加盟国およびEFTA加盟国全体において、各国の法律に移行する必要はなく直ちに発効されます。ただし、一部の要件をより詳細に裏付けるために、国内法が適応される場合があります。
機器のEU市場での継続的な供給を確保するために、十分な時間とリソースを提供することを主な目的として、4つの追加規制が制定されました。
| 規制 | 主要条項 |
| (EU) 2022/112 |
・レガシーデバイスに関する経過措置の第一次延長 |
| (EU) 2023/503 | ・ノーティファイドボディの再評価頻度の延長 |
| (EU) 2023/607 | ・移行期間中に上市された機器の売却期間の撤廃 |
| (EU) 2024/1860 | ・経過措置の第二次延長 ・IVDRに準拠した品質システムの実施に関するタイムラインの追加 ・ノーティファイドボディへの申請およびノーティファイドボディとの署名済み契約の完了に関するタイムラインの追加 ・欧州医療機器データベース(EUDAMED)の段階的な展開 ・予想される機器の供給中止を通知する義務の導入 |
以下の要件を満たす「レガシーデバイス」のみ、IVDR移行タイムラインが延長されます。
a) 当該機器が引き続き指令 98/79/EC に適合している
b) 設計および意図された目的に重要な変更がない
c) 当該機器が患者、ユーザーまたはその他の者の健康もしくは安全、または公衆衛生保護の側面に対して容認できないリスクをもたらさない
移行期間は、機器の分類と、自己宣言によってIVDDに基づいてCEマークが付けられたか、またはノーティファイドボディによる認証によってCEマークが付けられたかによって異なります。以下のタイムラインが適用されます:
下記の期日までにノーティファイド・ボディ(NB)に申請する必要があります。その後、各年の9月26日までにNBと契約を締結する必要があります。機器のクラスに応じて、以下に示す移行期間の終了まで、製品を市場に継続して提供することができます。
すべてのレガシーデバイスについては、IVDRの適用日(2022年5月26日)から第110条(3)に基づき、IVDR規則の一定の要件(市場後監視、ビジランス、エコノミックオペレーターの登録、市場監視など)が適用されます。
以下のインフォグラフィックで、IVDR移行規定のタイムラインの概要をご確認ください:

テュフズードは、EUの指令および規則で対象となるあらゆる種類の医療機器に対する世界最大級のEUノーティファイドボディの一つであり、IVDR規則に基づくフルスコープのノーティファイドボディとに指定されています。 当社は専門家の経験と専門知識により、EU市場に体外診断用医療機器を上市するために必要な認証取得の信頼できるサービスプロバイダーとなり、グローバル市場アクセスのための幅広い認証および試験サービスを提供します。
当社はパートナーと協力して、患者および医療従事者に重要で革新的な診断技術へのアクセスを確保します。
ほとんどの種類の医療機器の複雑な開発プロセスに加え、新しい規制要件に対応し、ノーティファイドボディの認証を得る必要があるため、ほとんどの医療機器製造業者にとって移行は複雑で時間のかかるプロセスになる可能性があります。さらに、以前に認証された機器であっても新しい規制の要件から免除されるわけではなく、再評価および再認証が必要です。
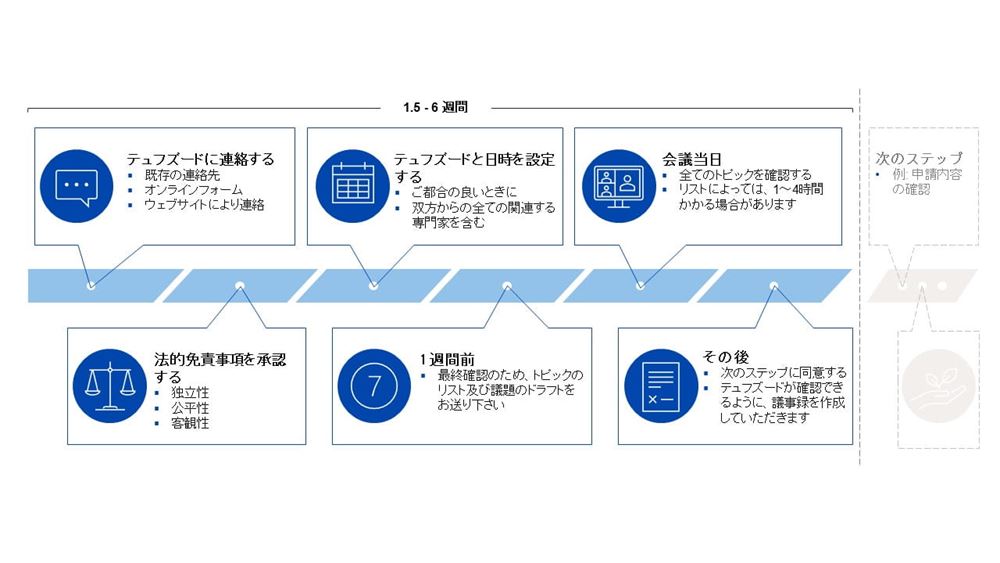
テュフズードに正式なIVDR申請を提出する前にストラクチャード・ダイアログを行う目的は、申請プロセスとフォームおよび提出書類のタイミング、手続き、規制の側面を明確にすることです。
ストラクチャード・ダイアログは、適合性評価の申請前に当社と会う機会であり、評価とは独立しています。
以下で、可能なトピック、プロセス、およびストラクチャード・ダイアログの申請方法について確認してください。
申請及びオンボーディング
製造業者のトピック
適合性評価手順
テュフズードノーティファイドボディの既存のお客様:
テュフズードへの申請を検討中のお客様:
製品のクラス分類に基づいて、製造業者は該当する適合性評価手順を申請する必要があります。
申請手続きを開始するにあたり、簡単なサービスリクエストフォームを使用して当社に連絡ください。
ご注意ください:TÜV SÜD Product Service GmbH は、IVDR 附属書 X に基づく適合性評価サービスを提供しておりません。IVDR 附属書 XI に基づくクラス C および D の機器の申請時には、対象機器の適合性評価活動に指定されたノーティファイドボディが発行した EU 型式審査認証書(EU Type-Examination Certificate)のコピーを製造業者が提出する必要があります。
書類の提出および関連する対応で使用できる言語は、英語またはドイツ語のみです。
認証費用は時間単価に基づいています。これには、企業の規模、サイトの数、機器の数およびその複雑さなどの要素が考慮されます。
TÜV SÜD Product Service GmbH よって提供される適合性評価活動の標準料金は以下の通りです:
| サービス | 時間単価* |
| 審査および品質マネジメントシステムアセスメントサービス | |
| 審査 | 350 €/時間 |
| 品質マネジメントシステム(IVDR)の変更通知と拡張の評価 | 350 €/時間 |
| 技術文書評価サービス | |
| オフサイト技術文書評価 | 465 €/時間 |
| 臨床評価 | 465 €/時間 |
| アプリケーション管理手数料 |
|
| ケースあたり | 2,800 € |
| ビジランス情報の初期評価 |
|
| 各ケース数 1-200 |
420 € |
| 各ケース数 > 200 |
90 € |
*製造業者の所在地や、適合性評価手続きに現地の専門家および審査員を含めた場合、価格が変動する可能性があります。また料金は現地通貨で請求されることがあります。
IVDRへの準拠するプロセスを円滑に進めるために、以下のリソースをご利用ください。
IVDR 品質マネジメントシステム要求事項
このチェックリストは、製造業者と審査チームが要求事項が満たされている場所や説明されている場所を確認するのに役立ちます。
IVDR技術文書の提出要求事項
このチェックリストは、ノーティファイドボディに提出するための技術文書を準備するのに役立ちます。
IVDR サンプリング
テュフズードが IVDR におけるクラス B 及びクラス C 機器のサンプリングをどのように実施しているかを理解します。実務上の補足説明とその意味を確認してください。
IVDRにおけるレガシー製品
体外診断用医薬品指令(IVDD)下で既に上市されている製品がIVDRに移行する方法についての情報。
欧州体外診断用医療機器規則(IVDR)
このインフォシートでは、IVDR の概要と必要事項を説明します
IVDR クラス D
このインフォシートでは、高リスクのクラス D 機器に対する特別な審査の規定に関する情報を提供します。
IVDR 分類
このインフォシートでは、IVDR(EU)2017/746に基づく医療機器のクラス分類に関する情報を提供します。


IVD機器向けのEMC試験個別規格の最新版「IEC 61326-2-6:2020」が発行され、試験の考え方が旧規格より大幅に変更されました。
もっと知る


国内薬機法において、すべての能動型医療機器は2023年3月より「JIS T 0601-1-2:2018(Ed.4.0)」に適合する必要があります。
もっと知る

Site Selector
Global
Americas
Asia
Europe
Middle East and Africa