
So erschließen Sie den EU-Markt für In-vitro-Diagnostika (IVD)
So erschließen Sie den EU-Markt für In-vitro-Diagnostika (IVD)
Die Verordnung über In-vitro-Diagnostika (IVDR 2017/746) ist die derzeit geltende Gesetzgebung der Europäischen Union (EU) für das Inverkehrbringen, die Bereitstellung und die Inbetriebnahme von In-vitro-Diagnostika auf dem europäischen Markt.
Die IVDR wurde am 5. Mai 2017 im Amtsblatt der EU veröffentlicht, trat am 26. Mai 2017 in Kraft und ersetzt schrittweise die frühere EU-Richtlinie über In-vitro-Diagnostika (98/79/EG).
Als EU-Verordnung ist die IVDR in allen Mitgliedstaaten der EU und der EFTA rechtskräftig. Eine Umsetzung in nationales Recht ist nicht mehr erforderlich. Nationale Gesetze können jedoch angepasst werden, um bestimmte Anforderungen näher zu konkretisieren.
Hier für IVDR-Serviceanfrage registrieren (engl.)
Die IVDR-Servicebeschreibung zum Download finden Sie hier.
Vier zusätzliche Verordnungen wurden erlassen, um die fortlaufende Verfügbarkeit von Produkten auf dem EU-Markt sicherzustellen und ausreichend Zeit und Ressourcen bereitzustellen.
| Verordnung | Wichtigste Bestimmungen |
| (EU) 2022/112 |
|
| (EU) 2023/503 |
|
| (EU) 2023/607 |
|
| (EU) 2024/1860 |
|
Die Übergangsfristen der IVDR gelten nur für Bestandsprodukte, die die folgenden Bedingungen erfüllen:
a) Die Produkte müssen auch weiterhin der Richtlinie 98/79/EG entsprechen.
b) Es liegen keine wesentlichen Änderungen der Auslegung und der Zweckbestimmung vor.
c) Die Produkte stellen kein unannehmbares Gesundheits- oder Sicherheitsrisiko für Patient*innen, Anwender*innen, andere Personen oder für andere Aspekte des Schutzes der öffentlichen Gesundheit dar.
Die Übergangsfrist richtet sich nach der künftigen Risikoklasse eines Produkts und danach, ob die IVDD-Konformitätserklärung durch eine Benannte Stelle oder durch den Hersteller ohne Beteilung einer Benannten Stelle ausgestellt worden ist. Es gelten folgende Fristen:
• 26. Mai 2025 – Umsetzung eines IVDR-konformen Qualitätsmanagementsystems (QMS)
• 26. Mai 2025 – Antrag auf Konformitätsbewertung bei einer Benannten Stelle
• 26. September 2025 – Abschluss einer schriftlichen Vereinbarung zwischen einer Benannten Stelle und dem Hersteller
• 31. Dezember 2027– Ende der Übergangsfrist
• 26. Mai 2025 – Umsetzung eines IVDR-konformen QMS
• 26. Mai 2025 – Antrag auf Konformitätsbewertung bei einer Benannten Stelle
• 26. September 2025 – Abschluss einer schriftlichen Vereinbarung zwischen einer Benannten Stelle und dem Hersteller
• 31. Dezember 2027 – Ende der Übergangsfrist
• 26. Mai 2025 – Umsetzung eines IVDR-konformen QMS
• 26. Mai 2026 – Antrag auf Konformitätsbewertung bei einer Benannten Stelle
• 26. September 2026 – Abschluss einer schriftlichen Vereinbarung zwischen einer Benannten Stelle und dem Hersteller
• 31. Dezember 2028 – Ende der Übergangsfrist
• 26. Mai 2025 – Umsetzung eines IVDR-konformen QMS
• 26. Mai 2027 – Antrag auf Konformitätsbewertung bei einer Benannten Stelle
• 26. September 2027 – Abschluss einer schriftlichen Vereinbarung zwischen einer Benannten Stelle und dem Hersteller
• 31. Dezember 2029 – Ende der Übergangsfrist
• 26. Mai 2025 – Umsetzung eines IVDR-konformen QMS
• 26. Mai 2027 – Antragstellung bei einer Benannten Stelle
• 26. September 2027 – Abschluss einer schriftlichen Vereinbarung zwischen einer Benannten Stelle und dem Hersteller
• 31. Dezember 2029 – Ende der Übergangsfrist
• Für diese Produkte gilt keine Übergangsfrist – sie müssen seit dem 26. Mai 2022 vollständig der IVDR entsprechen.
Ihren Antrag bei der Benannten Stelle müssen Sie spätestens zu den genannten Terminen stellen. Als Nächstes müssen Sie bis zum 26. September des jeweiligen Jahres einen Vertrag mit einer Benannten Stelle abschließen. Ihre Produkte können Sie dann, je nach Produktklasse, bis zum Ende der unten aufgeführten Übergangsfristen weiterhin in Verkehr bringen .
Für alle Bestandsprodukte gelten gemäß Artikel 110 (3) seit dem 26. Mai 2022, also seit Geltungsbeginn der IVDR, bestimmte Anforderungen wie die Überwachung nach dem Inverkehrbringen, die Marktüberwachung, Vigilanz, Registrierung der Wirtschaftsakteure und Produkte.
Die nachfolgende Infografik zeigt alle Fristen für Übergangsbestimmungen auf einen Blick:

Stellen Sie sicher, dass Ihr QMS, den in Artikel 10 (8) der IVDR festgelegten Anforderungen entspricht.
Erstellen Sie Ihre Technische Dokumentation gemäß der Anhänge II und III der IVDR:
Wenden Sie sich frühzeitig an uns, um Ihre Bedürfnisse zu besprechen:
Bleiben Sie stets über die neuen Verordnungen und Leitlinien auf dem Laufenden:
TÜV SÜD ist eine der weltweit größten Benannten Stellen für alle Arten von Medizinprodukten, die in den Geltungsbereich von EU-Richtlinien und EU-Verordnungen fallen, und eine Benannte Stelle mit vollem Geltungsbereich (Full Scope Notified Body) unter der IVDR.
Mit unserer Erfahrung und unserem Fachwissen sind wir Ihr Partner des Vertrauens für das Inverkehrbringen Ihrer In-vitro-Diagnostika auf dem europäischen Markt und alle diesbezüglich benötigten Zertifizierungen. Wir bieten die gesamte Palette an Prüfdienstleistungen und Zertifizierungen für den weltweiten Marktzugang. Gemeinsam mit unseren Partnern stellen wir sicher, dass Patient*innen und Anbieter*innen auch künftig Zugang zu entscheidenden innovativen Diagnosetechnologien haben.
Da der Entwicklungsprozess bei den meisten Medizinprodukten komplex ist und Hersteller sich mit den neuen regulatorischen Anforderungen auseinandersetzen und die Zustimmung der Benannten Stellen einholen müssen, gehen Fachkreise davon aus, dass die Umstellung auf die neue Verordnung für die meisten IVD-Hersteller kompliziert und zeitaufwendig wird. Die Anforderungen der neuen Verordnung gelten auch für bereits zugelassene Bestandsprodukte. Auch diese müssen neu bewertet und zugelassen werden.
Der Strukturierte Dialog erfolgt vor Einreichen des offiziellen IVDR-Antrags bei TÜV SÜD. Im Dialog sollen der zeitliche Ablauf, das Verfahren und die regulatorischen Aspekte des Antragsprozesses geklärt sowie die Fragen bezüglich der Antragsformulare und der einzureichenden Unterlagen beantwortet werden.
Der Strukturierte Dialog bietet Ihnen die Möglichkeit, sich vor Einreichen des Antrags auf Konformitätsbewertung mit uns zu treffen, und ist von dieser völlig unabhängig.
Klappen Sie die Registerkarten unten auf und erfahren Sie mehr über die möglichen Themen, den Ablauf und die Beantragung eines Strukturierten Dialogs.
Antrag und Onboarding
• Onboarding Prozess
• Antragsformulare und Antragsprüfungsprozess
• Standorte, Lieferanten und Produkte
• Produktklassifizierung und Code-Zuweisung
Fragen der Hersteller
• TÜV SÜD Prüf- und Zertifizierungsordnung
• IVDR-Rahmenvereinbarung
• Umgang mit dem Medizinprodukt und mögliche Änderungen
• Wechsel zu TÜV SÜD als Benannte Stelle
Konformitätsbewertungsverfahren
• Projektplanung, Fristen für besondere Verfahren (z. B. Konsultationen)
• Vorlagepflichten
• Kosten, Gebühren und andere wirtschaftliche Aspekte
• Gemeinsame Spezifikationen, Leitfäden und harmonisierte Normen

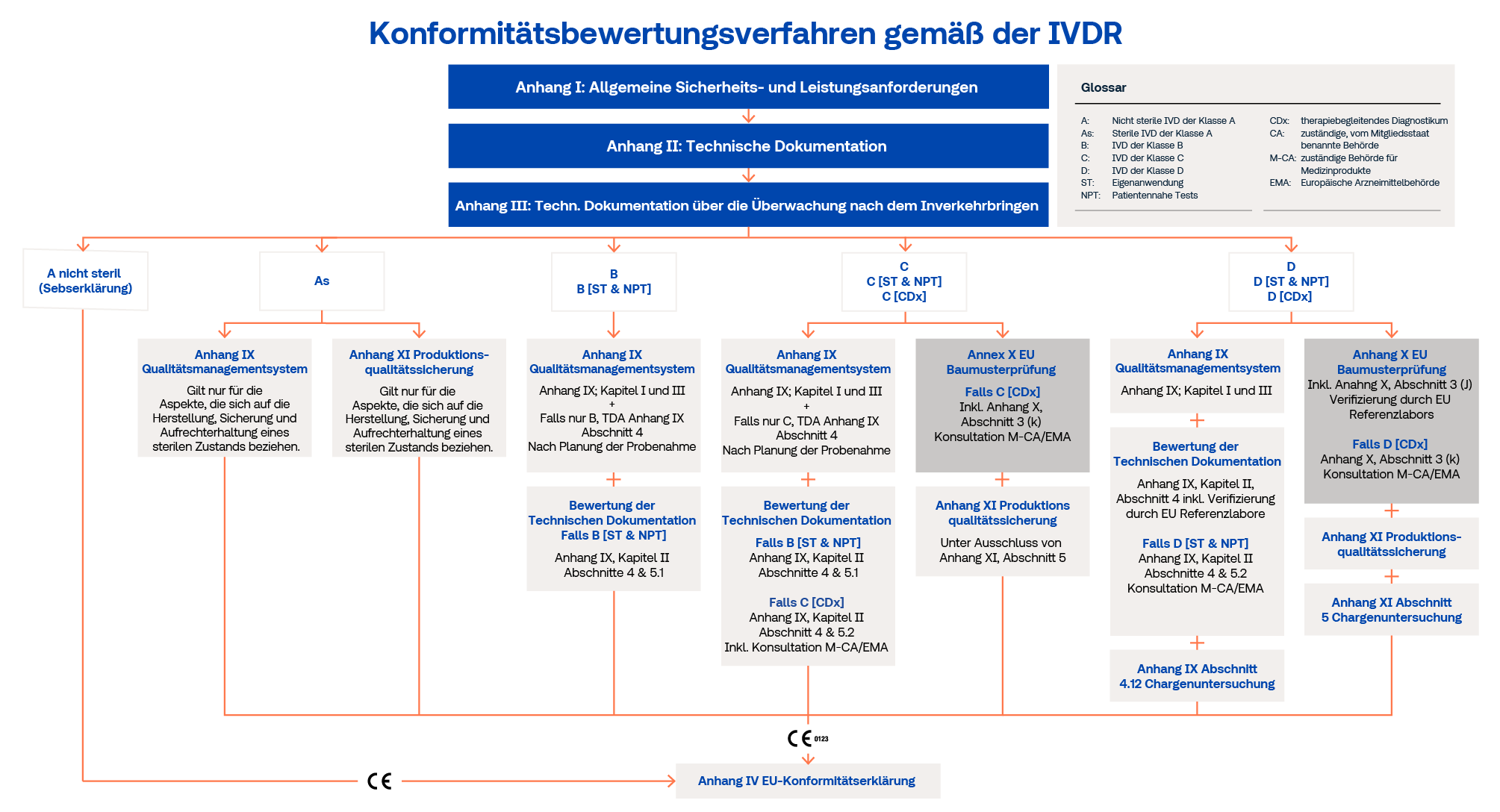
Der Hersteller muss ausgehend von der Produktklassifizierung das für sein Produkt geltende Konformitätsbewertungsverfahren beantragen.
Kontaktieren Sie uns einfach mithilfe unseres Service-Antragsformulars und starten Sie Ihren Antragsprozess.
Die TÜV SÜD Product Service GmbH führt keine Konformitätsbewertungen nach IVDR Anhang X durch. Bei der Einreichung eines Antrags gemäß IVDR-Anhang XI für Produkte der Klassen C und D muss der Hersteller eine Kopie der EU-Baumusterprüfbescheinigungen vorlegen, die von einer benannten Stelle ausgestellt wurden, die für Konformitätsbewertungstätigkeiten in Bezug auf die betreffenden Produkttypen benannt wurde.
Die eingereichte Dokumentation und die dazugehörige Korrespondenz müssen in Deutsch oder Englisch abgefasst sein.
Die Zertifizierungskosten berechnen sich auf der Basis von Stundensätzen und berücksichtigen Faktoren wie die Größe des Unternehmens, die Anzahl der Standorte und die Zahl und Komplexität der Produkte.
Die TÜV SÜD Product Service GmbH berücksichtigt die Interessen kleiner und mittlerer Unternehmen (KMU), indem sie ihre gesamte Gebührenstruktur vorrangig an aufwandsbezogenen Kriterien und nicht an festen Gebührenstufen ausrichtet. Dies ermöglicht insbesondere KMUs eine genaue und individuelle Kostenkalkulation auf Basis des jeweiligen Aufwands pro Einzelfall.
Für von der TÜV SÜD Produkt Service erbrachte Konformitätsbewertungsaktivitäten werden folgende Standardgebühren erhoben:
| Dienstleistung | Stundensatz* |
| Dienstleistungen im Bereich Auditierung und QMS-Bewertung | |
| Audit | 350 EUR |
| Bewertung von Änderungsmeldungen und Verlängerungen für Qualitäts- und Sicherheitsmanagementsysteme, IVDR | 350 EUR |
| Prüfdienstleistungen für Technische Dokumentation | |
| Offsite-Bewertung der Technischen Dokumentation | 465 EUR |
| Klinische Bewertung | 465 EUR |
| Gebühr für das Antragsmanagement | |
| Pro Fall | 2.800 EUR |
| Erstbewertung von Vigilanzinformationen | |
| 1 bis 200 Fälle | 420 EUR |
| Mehr als 200 Fälle | 90 EUR |
*Die Preise können abhängig vom Standort des Herstellers und der Möglichkeit, lokale Experten oder Auditoren in das Konformitätsbewertungsverfahren einzubeziehen variieren; Gebühren können auch in der Landeswährung in Rechnung gestellt werden.
Sorgen Sie für einen reibungslosen Prozess zur Einhaltung der IVDR mit den folgenden Ressourcen.
| Auditcheckliste | |||
|
IVDR-Anforderungen an QM-Systeme Mit dieser Checkliste können sich Hersteller und Audit-Teams auf das Audit vorbereiten und feststellen, wo Anforderungen erfüllt oder beschrieben sind.
|
|
||
| Zertifizierungsleitfaden | |||
|
Erweiterte IVDR-Übergangsfristen
Hier finden Sie einen kurzen Leitfaden zu den IVDR-Fristen:
|
Stichprobenplan für die Bewertung der Technischen Dokumentation von Klasse-B- und-C-Produkten Erfahren Sie, wie TÜV SÜD diese Anforderungen umsetzt. Weitere praktische Erklärungen und Implikationen finden Sie hier: |
IVDR-Anforderungen für Bestandsprodukte Hier finden Sie mehr Informationen darüber, wie Produkte, die bereits unter der In-vitro-Diagnostika-Richtlinie 98/79/EG (IVDD) auf dem Markt sind, auf die IVDR umgestellt werden können. |
Erfahren Sie, wie Sie die IVDR-Zertifizierung für CDx-Produkte erfolgreich abschließen können. |
| Zusätzliche Informationen | |||
|
Die Verordnung der Europäischen Union über In-vitro Diagnostika |
|||
|
IVDR Klasse D |
|||
|
IVDR-Klassifizierung |
|||
IVDR - Welche Prüfnachweise sind für Hersteller von Medizinprodukten erforderlich und auf was müssen sie achten?
Jetzt reinhören!